Cisztás fibrózis
Szinonimák tágabb értelemben
Cisztás fibrózis, tüdő
Angolul: mucoviscidosis, cisztás fibrózis
A cisztás fibrózis meghatározása
A cisztás fibrózis öröklött betegség. Az öröklődést orvosi szempontból autoszomális recesszívnek nevezik. A cisztás fibrózist (cisztás fibrózist) nem az X és Y nemi kromoszómán, hanem az autoszomális 7. kromoszómán öröklik.
Olvassa el az anyagcsere-rendellenességekre vonatkozó általános cikket: Anyagcsere-rendellenességek - mit jelent ez?
A mutáció az úgynevezett CFTR génen található. A recesszív azt jelentette, hogy a betegség kitöréséhez a génnek két hibás másolatának kell lennie. Ha egy személy egészséges és mutáns génhelyzettel rendelkezik a megfelelő 7. kromoszómán, akkor a betegség nem fordul elő.
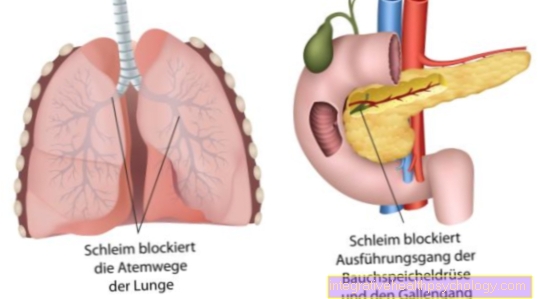
Az eredmény patológiás géntermék. Az így kódolt Klorid csatornák megtört. A hibás kloridcsatornák vastag nyálka kialakulásához vezetnek az összes exokrin mirigyben.
Ezek az exokrin mirigyek, azaz olyan mirigyek, amelyek felszabadítják szekréciójukat kívülről, a következők:
- a hasnyálmirigy
- a vékonybél
- légúti rendszer tüdővel és hörgőkkel
- epevezeték és
- szintén a Verejtékmirigyek
összefoglalás
Cisztás fibrózis egy Örökletes betegség. Olyan módon örököl, hogy nemtől független, és csak vele együtt két hibás gén bekövetkezik. Ez a leggyakoribb autoszomális recesszív öröklés.
A következmények: kemény nyálkahártya-képződmények az összes exokrin mirigy, mint például a tüdő mirigyek, a hasnyálmirigy és az izzadságok. Ezen alapulnak zavart a klorid szállítása a cella belső és külső része között (olvassa tovább: Klorid a vérben). A mutáns gén be van kapcsolva 7. kromoszóma és a szervek sokféle részvételét okozza, ennek megfelelő hatása van a légzésre, az emésztésre és a szaporodásra.
Sajnos a terápia csak enyhítheti a tüneteket, de nem eredményez gyógyulást. A Várható élettartam cisztás fibrózisban szenvedő betegekben viszonylag alacsony.
Mivel a betegséget recesszív módon öröklik, vannak olyan emberek, akik hordozzák a módosított gént, de nem szenvednek a betegségtől. Ilyen személyeket hívnak Funkcióhordozó vagy Vezetékek, azaz hordozók. Ezeknek az embereknek nincs cisztás fibrózisa, mert a gén másik példánya ép és a beteg nem elég erős ahhoz, hogy uralkodjon.
A gén ezen hibás másolatát azonban továbbadhatja utódai számára. Ha egy módosított gén már elegendő egy betegség kiváltásához, akkor az úgynevezett domináns örökség lenne. Ilyen örökség megtalálható például a Chorea Huntington. A betegségről többet tudhat meg a téma alatt Chorea Huntington.
Körülbelül 1:2500 fekszik a Betegség aránya újszülöttekben Németországban. Hordozó mindenkinek szól 25. a német lakosságban.
kiváltó ok

A cisztás fibrózist egy gén mutációja okozza a 7. kromoszómán. Ez a kromoszóma egy autoszomális kromoszóma, nem pedig a nemi kromoszóma.
Mindenkinek van 44 autoszomális kromoszóma (mindegyik két azonos verziója) és két nemi kromoszóma. Ez a mutáció a 7. kromoszómán hibás klorid csatornák kialakulásához vezet. A klorid reabszorpciója (reabszorpciója) a mirigyszekréciókból nem lehetséges, mivel a receptor, a klorid dokkoló pontja nem épül be a mirigycsatornákba.
Ehelyett helytelen megjelenés és felépítés miatt bányászatra bontják. Megzavarja a klorid természetes cseréjét bizonyos klorid csatornákon keresztül. Ezek az úgynevezett csatornák fehérjékből állnak. A fehérjék széles skáláját kódoljuk a DNS-en. A klorid csatornák genetikai hibája miatt az összes mirigyből kiszáradt és kemény nyálkahártya képződik, amelyek szabadon engedik a szekréciót. A nyálka ezután részlegesen elzárja a légcsöveket vagy a tüdőben lévő légutakat.
Olvassa el erről is Kromoszóma mutáció
Cisztás fibrózis diagnosztizálása

A csecsemőkorban kezdődő tipikus tünetek úttörő szerepet játszanak a cisztás fibrózis diagnosztizálásában.
Ezt a gyanút megerősíti a pozitív családi anamnézis (az apa / anya vagy közeli hozzátartozóinak betegsége). A pozitív családi kórtörténet azt jelenti, hogy a családon belül vannak vagy már voltak cisztás fibrózis esetek - az anya vagy az apja oldalán.
A hasnyálmirigy enzimek hiánya kimutatható a székletben is. A légutak bármilyen eltömődését a mellkas röntgenfelvétele segítségével lehet észlelni.
A verejték tesztje, amely az izzadság klorid-tartalmát méri, szintén segít a cisztás fibrózis diagnosztizálásában. Ha egy értéket túllépnek, és a többi tünet szintén érvényes, a diagnózis viszonylag rögzített. A szülők gyakran maguk észlelik a megnövekedett sótartalmat a csecsemő verejtékében.
A születendő gyermeket ezen örökletes betegség szempontjából is meg lehet vizsgálni. Amniotikus folyadék szúrással (magzatvíz mintavétel) a magzati sejteket eltávolítottuk és megvizsgáltuk a mutált gént.
További információ a témáról: A gyermek röntgen vizsgálata
Cisztás fibrózis kezelése
A cisztás fibrózisban szenvedő személyek egyben kapnak tanácsot Cisztás fibrózis - járóbeteg részleg vagy tanácsot Emberi genetikus (Örökletes betegségek szakember) ajánlott. Ezek elősegíthetik az életminőség javítását, vagy ha gyermeket akarnak, akkor kiszámíthatják a beteg gyermek valószínűségét. Feltéve, hogy a szülők termékenyek és termékenyek.
Egyébként a kezelés tüneti, mivel az okot, a hibás gént, nem lehet kiküszöbölni.
Gyógyíthatatlan betegség
A cisztás fibrózis (cisztás fibrózis) ma is gyógyíthatatlan betegség.
Cisztás fibrózis esetén fontos, hogy megfelelő mennyiségű étkezési sót (Nátrium-klorid, NaCl). A mukolízis természetesen célja. A mukolízis a nyálka feloldódása, különösen a tüdőben, hogy megkönnyítse a légzést.
A gyógyszerek és az inhaláció enyhíthetik a tüneteket. Ha a tüdő működése észrevehetően romlik, akkor oxigén adható.
Intenzív fizikoterápián keresztül (fizikoterápia), például a masszázs és a légzés gyakorlása révén a cisztás fibrózis által okozott tüdőváltozásokat is kezelik.
Gyakran előfordul, hogy a betegség a szükséges tüdőátültetéssel zárul le. A várakozási listák azonban hosszúak.
A hasnyálmirigy enzimek és zsírban oldódó vitaminok szájon át történő beadása szintén a kezelés része. A hasnyálmirigy feladatát ezért támogatni kell, vagy inkább helyettesíteni kell. A zsírban oldódó vitaminok A, D, E és K. Ezeket közvetlenül a vérbe kell adni, mivel emésztő enzimek hiánya miatt nem képesek felszívódni az élelmiszerekből.
Az étrendnek magas kalóriatartalmúnak is kell lennie, mivel ezeknek csak töredéke nyerhető élelmezésből.
A szövődmények, például az influenza vagy tüdőgyulladás további kockázati tényezőinek elkerülése érdekében a gyermeket oltani kell. A következő oltások ajánlottak:
- kanyaró
- pneumococcusokat
- influenza
További információ a témáról: Felülfertőződés
Ezekhez az intézkedésekhez természetesen orvoshoz kell fordulni, akivel meg kell beszélni a kockázatokat.
Manapság a cisztás fibrózis terápia nagy reményét helyezi a genetikai kutatás. Kísérlet történik a hiányzó genetikai információ bevezetésére az emberi genomba. Olyan vektorokat keresünk, amelyek képesek ezt a feladatot elsajátítani. A vektorok lehetnek például baktériumok vagy vírusok, amelyek képesek beépíteni az egészséges gyakoriságot genetikai felépítésünkbe.
A születendő betegek terápiás megközelítését jelenleg tesztelik. Egerekben az egér embrióknak már sikerült bevezetni az egészséges gént, amely tartalmazza a helyes génszekvenciát, amniocentesis (amniotikus folyadék oltás) útján. Ilyen egerekben az egészséges CFTR gén termelődött. Az amniocentesis a gyermek sejtjeinek szúrása és eltávolítása az amniotikus folyadékból, amelyet az anya hasfalán keresztül végeznek.
Németországban azonban az intrauterin (= méhben = a méhben) "terápia" e formája tilos.
megelőzés
A megelőző intézkedés ebben az értelemben nem létezik, mert örökletes betegség.
Ugyanakkor meglátogatható egy emberi genetikai tanácsadó központ (általában az egyetemi kórházakban található). Itt kiszámítják, hogy mekkora a kockázata annak, hogy a betegséget átadják a gyermekeknek.
Ez a tanács mindig hasznos, ha a családban kórtörténetében van cisztás fibrózis.
Egy is Prenatális diagnosztika érdemes törekedni. Itt a szülés előtt (azaz prenatálisan) a Amniotikus folyadék vizsgálata (magzatvíz mintavétel) végzett. A magzati sejteket (a gyermek sejtjeit) vesszük az amniotikus folyadékból, és megvizsgáljuk a DNS-t a mutáns gén szempontjából.
A cisztás fibrózis előrejelzése
Sajnos a cisztás fibrózisban szenvedő betegek átlagos élettartama csak 32-37 év. Manapság az ilyen állapotban született újszülöttek várható élettartama körülbelül 45-50 év.
A prognózis nagyban függ a terápiától és annak betartásától.
Ezért a beteg és motivációja fontos szerepet játszik.